
In recent years, the tumor microenvironment has gained as much importance as the tumor itself. Some tumors are considered “inflamed” (that is, rich in activated T cells that recognize and attack malignant cells) and tend to respond better to treatments like immune checkpoint inhibitors (e.g., anti–PD-1). However, most solid tumors in patients do not exhibit this immune activation: they are “non-inflamed” tumors, characterized by a scarcity of T cells, an abundance of immunosuppressive macrophages, and a poor response to immunotherapy. What determines this immune profile? In this new study, the research team set out to understand why some tumors manage to remain “silent” to the immune system, even when they carry mutations that should trigger a defense response.
Using spontaneous hepatocellular carcinoma models in mice—with specific mutations that mimic subtypes of human liver cancer—the researchers identified a key factor: non-inflamed tumors secreted large amounts of erythropoietin (EPO), a hormone primarily known for its role in red blood cell production. This finding was unexpected, as EPO is not usually associated with tumor immunology. Most interestingly, this production of EPO was not related to anemia, indicating that it was not a normal physiological response, but rather a tumor strategy. Indeed, when analyzing clinical data from patients with hepatocellular carcinoma, they found that high EPO expression in the tumor was associated with poorer prognosis, greater vascular invasion, and lower cellular differentiation.
The key mechanism lies in the EPO receptors (EPOR), which are abundantly expressed on tumor-associated macrophages (TAMs), particularly in a subtype resembling Kupffer cells—the liver’s resident macrophages. When EPO binds to its receptor on these cells, it reprograms them to adopt an immunosuppressive profile: they reduce their capacity for antigen presentation (e.g., lowering expression of major histocompatibility complex class II), decrease production of pro-inflammatory cytokines, and promote the accumulation of regulatory T cells (Tregs), which inhibit the activity of cytotoxic T cells. Altogether, this environment suppresses the immune attack on the tumor.
To test whether tumor-derived EPO was directly responsible for this immune profile, the researchers performed two types of experiments. In one, they genetically eliminated EPO production in non-inflamed tumors, which led to greater infiltration of CD8+ T cells, immune activation, and tumor regression. In the other, they forced EPO expression in tumors that were normally inflamed and observed that they quickly lost their immune-active profile and became resistant to immunotherapy. This demonstrated that EPO acts as a true “immunosuppressive switch” capable of determining the type of tumor microenvironment, regardless of the tumor’s genetic profile.
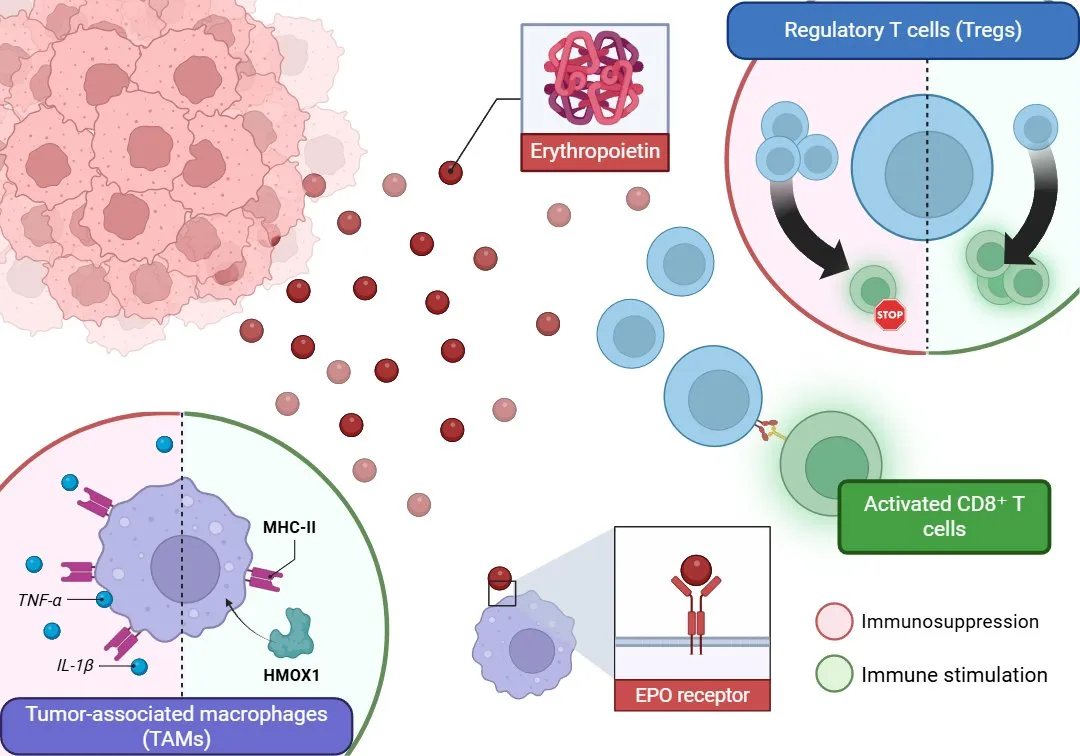
Figure 1. Tumor-derived erythropoietin (EPO) binds to its receptor (EPOR) on tumor-associated macrophages (TAMs), promoting an immunosuppressive phenotype via downregulation of MHC-II and proinflammatory cytokines (IL-1β, TNF-α), and upregulation of HMOX1. This reprogramming enhances regulatory T cell (Treg) activity and inhibits cytotoxic CD8⁺ T cells, leading to immune evasion.
The study also revealed that EPO’s effect on macrophages is mediated through a specific intracellular pathway involving the transcription factor NRF2. This molecule regulates genes involved in heme degradation and antioxidant production—both processes that suppress inflammation. In macrophages stimulated by EPO, a decrease in pro-inflammatory cytokines like IL-1β and TNF-α was observed, along with an increase in proteins such as HMOX1 (heme oxygenase), which degrade heme and generate a more tolerogenic environment. In mice genetically modified to lack EPOR or NRF2 in their macrophages, tumors not only grew more slowly but also responded effectively to anti–PD-1 immunotherapy, which typically fails in non-inflamed tumors.
This work not only changes our understanding of EPO’s role in cancer but also opens new therapeutic avenues. By blocking EPO/EPOR signaling—whether genetically, with pharmacological inhibitors, or with macrophage-targeted interfering RNA—the authors were able to transform “cold” (non-inflamed) tumors into “hot” (inflamed) ones, making them responsive to immunotherapy. This strategy could be applied not only in liver cancer but also in other solid tumors such as breast, kidney, or colon cancer, where EPO has also been associated with poor prognosis.
Main Reference:
Chiu, D. K., Zhang, X., Cheng, B. Y., Liu, Q., Hayashi, K., Yu, B., Lee, R., Zhang, C., An, X., Rajadas, J., Reticker-Flynn, N. E., Rankin, E. B., & Engleman, E. G. (2025). Tumor-derived erythropoietin acts as an immunosuppressive switch in cancer immunity. Science (New York, N.Y.), 388(6745), eadr3026. https://doi.org/10.1126/science.adr3026
Other References:
Galon, J., Costes, A., Sanchez-Cabo, F., Kirilovsky, A., Mlecnik, B., Lagorce-Pagès, C., Tosolini, M., Camus, M., Berger, A., Wind, P., Zinzindohoué, F., Bruneval, P., Cugnenc, P. H., Trajanoski, Z., Fridman, W. H., & Pagès, F. (2006). Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science (New York, N.Y.), 313(5795), 1960–1964. https://doi.org/10.1126/science.1129139
Galon, J., & Bruni, D. (2019). Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nature reviews. Drug discovery, 18(3), 197–218. https://doi.org/10.1038/s41573-018-0007-y
Kurtulus, S., Madi, A., Escobar, G., Klapholz, M., Nyman, J., Christian, E., Pawlak, M., Dionne, D., Xia, J., Rozenblatt-Rosen, O., Kuchroo, V. K., Regev, A., & Anderson, A. C. (2019). Checkpoint Blockade Immunotherapy Induces Dynamic Changes in PD-1-CD8+ Tumor-Infiltrating T Cells. Immunity, 50(1), 181–194.e6. https://doi.org/10.1016/j.immuni.2018.11.014